 science-review.ru
science-review.ru
Введение
Синдром Боринга – Опица (СБО) – редкое тяжелое аутосомно-доминантное генетическое заболевание, впервые описанное Bohring и соавт. в 1999 г. [1]. На сегодняшний день во всем мире описано 46 случаев СБО, большинство из которых были зарегистрированы в западных странах, 7 случаев – в азиатских странах [2, 3].
В 2011 г. группа ученых впервые обнаружила, что более чем у половины детей с СБО есть мутации в гене ASXL1, расположенном на длинном плече (q) хромосомы 20 в участке 20q11.21 [4]. В последующем вариант мутации в гене ASXL1 c.1210C>T/p.R404* (NM_015338.6) продемонстрировал очень убедительные доказательства патогенности [5, 6]. Генетическая мутация ASXL1 происходит при образовании яйцеклетки или сперматозоида и не унаследована от родителей. Такие мутации называются новыми или de novo – изменение в гене, произошедшее впервые и только у одного из членов семьи. Ген ASXL1 состоит из 13 экзонов и 12 интронов и кодирует нуклеопротеин длиной в 1541 аминокислоту, который широко экспрессируется в различных тканях, поэтому при СБО изменения могут возникать в разных органах [7, 8].
В 2011 г. R. Hastings и соавт. установили клинические диагностические критерии для этого заболевания, включающие микроцефалию, тригоноцефалию, гемангиому лица, типичную позу СБО, трудности с кормлением, задержку внутриутробного развития, отставание в психомоторном развитии и характерный черепно-лицевой порок развития [9]. Общие черепно-лицевые особенности включают выпуклые глаза, микро-/ретрогнатию и расщелину неба. У некоторых пациентов могут быть врожденные аномалии (структурные аномалии мозга, сердечные аномалии, аномалии опорно-двигательного аппарата и т.д.). На сегодняшний день зарегистрировано более 50 пациентов с СБО, соответствующих клинико-диагностическим критериям [10, 11].
У некоторых детей с синдромом СБО ген ASXL1 не изменен, что, вероятно, связано с изменениями в других связанных генах. Дифференциальная диагностика проводится с СБО-подобным фенотипом, связанным с геном KLHL7. Он имеет несколько общих черт с СБО (включая сходное положение верхних конечностей), но клинически отличается отсутствием тригоноцефалии, синопсиса, высокой миопии и циклической рвоты. Другие заболевания, имеющие несколько общих признаков с СБО, но лишенные осанки СБО, включают синдром С, синдром Шаши – Пена, синдром Бейнбриджа – Роперса и синдром Корнелии де Ланге. Функции гена и белка ASXL1 описаны не полностью, и в настоящее время продолжается изучение их особенностей, генетических причин СБО [12, 13].
Уровень младенческой смертности высок. У пациентов, переживших младенчество, трудности с кормлением и рецидивирующие инфекции становятся менее серьезными. У пожилых пациентов могут наблюдаться некоторые целенаправленные движения конечностей, однако в состоянии покоя они возвращаются к позе СБО. Умственная отсталость варьируется от тяжелой до глубокой.
Цель исследования – представить клинический случай синдрома Боринга – Опица и описать его клинические особенности.
Материалы и методы исследования
Представлен клинический случай ребенка И., госпитализированного в паллиативное отделение Городской детской клинической больницы № 17 (ГДКБ № 17) Уфы. Было проведено изучение медицинской документации, общеклиническое обследование, проанализированы результаты дополнительных исследований. Мать ребенка подписала добровольное информированное согласие на публикацию результатов обследования и лечения, фотографий ребенка в научно-практическом журнале.
Результаты исследования и их обсуждение
Анамнез жизни. Ребенок от 1 беременности, 1 родов. Беременность протекала на фоне хронической плацентарной недостаточности, нарушения маточно-плацентарного кровотока IА степени, многоводия, компенсированной гипоксии плода. Носитель вируса гепатита В (HBsAg), первичное бесплодие 10 лет. Беременность методом экстракорпорального оплодотворения с интрацитоплазматической инъекцией сперматозоида (ЭКО+ИКСИ). Врожденный порок сердца (ВПС) плода: дефект межжелудочковой перегородки. Роды при сроке 39,2 недель в головном предлежании. Масса тела при рождении 2850 г, рост 50 см, окружность головы 34 см. Оценка по шкале Апгар 7–8 баллов. Состояние ребенка после рождения тяжелое, обусловлено нарастанием дыхательных нарушений, перенесенной гипоксией. К груди не приложен по тяжести состояния. Предварительный диагноз. Основной: Дыхательное расстройство неуточненное. Дыхательная недостаточность (ДН) 1 ст. Сопутствующий: Задержка внутриутробного развития, внутриутробная гипотрофия 1 степени, врожденный порок сердца (ВПС): дефект межжелудочковой перегородки (установлен антенатально).
Анамнез заболевания. В первые часы жизни у ребенка наблюдалось резкое ухудшение состояния, обусловленное синдромом дыхательных расстройств. В первые сутки жизни переведен в отделение реанимации и интенсивной терапии Республиканского клинического перинатального центра. Проведено обследование: анализ крови на группу и резус-фактор – ВIII Rh+; клинический анализ крови, биохимический анализ крови, коагулограмма, общий анализ мочи – в пределах возрастной нормы; анализ крови методом ПЦР на вирус простого герпеса (ВПГ), цитомегаловирус (ЦМВ) – отрицательно; анализ крови на TORCH-инфекцию – обнаружены IgG к ЦМВ, ВПГ 1,2, краснухе; ультразвуковое исследование (УЗИ) головного мозга – гипоксически-ишемическое поражение центральной нервной системы (ЦНС); УЗИ брюшной полости, тимуса – патологии не выявлено; электрокардиограмма – синусовая тахикардия, ЧСС 171 уд/мин, электрическая ось сердца отклонена вправо; эхокардиография (ЭХО-КГ) – открытый артериальный проток; рентгенография органов грудной клетки (ОГК) – мелкоочаговые тени на фоне сгущения легочного рисунка в апикальных и медиальных отделах; бактериальный посев трахеального аспирата, кала – микроорганизмы не выделены. Выставлен диагноз: Врожденная пневмония неуточненная, острое течение, тяжелая форма. ДН у новорожденных III степени. Осложнения: «Маловесный» для гестационного возраста плод, задержка внутриутробного развития плода по гипотрофическому типу легкой степени тяжести. Контакт с больным и возможность заражения вирусным гепатитом В. Гипоксически-ишемическая энцефалопатия новорожденного. ВПС: открытый артериальный проток. В отделении проводилась этиотропная, патогенетическая, симптоматическая терапия.
На 7-е сутки жизни ребенок переведен в отделение анестезиологии и реанимации Республиканской детской клинической больницы по линии санавиации с ведущими синдромами: ДН по смешанному типу, неврологические нарушения, метаболические нарушения у доношенного новорожденного с гестационным возрастом 39 недель на фоне инфекционно-воспалительного процесса. С 8-е по 17-е сутки жизни находился на аппарате искусственной вентиляции легких (ИВЛ). Учитывая отсутствие положительной динамики, сохраняющуюся тяжелую ДН, проведена бронхоскопия, консультация оториноларинголога. Выставлен диагноз: Врожденный порок развития (ВПР): пролапс мембранозной части трахеи, бронхомаляция среднедолевого бронха справа. Ребенок оставлен на эндотрахеальной трубке, на ИВЛ. В возрасте 18 суток, учитывая длительное нахождение на ИВЛ, несколько неудачных попыток экстубации, по данным бронхоскопии – пролапс мембранозной части трахеи, бронхомаляция среднедолевого бронха справа, принято решением консилиума о наложении трахеостомы. Проведена операция – нижняя трахеостомия. В послеоперационном периоде ребенок находился на аппарате ИВЛ через трахеостому. Наблюдалось затруднение глотания, срыгивания, рвота, в связи с чем был установлен назогастральный зонд. Проведено обследование: анализ крови на HbsAg – отр., анализ крови на антитела к вирусу гепатита В – отр.; рентгенография ОГК в прямой проекции – пневмопатия; компьютерная томография ОГК с ангиографией – патологии не выявлено; электроэнцефалография – типичной эпиактивности и паттернов приступов не зарегистрировано; магнитно-резонансная томография головного мозга: признаки гипоксически-ишемического поражения головного мозга, церебральной незрелости, дисгенезия мозолистого тела, гипоплазия нижнего червя мозжечка с заместительным расширением субарахноидальных пространств задней черепной ямки; ЭХО-КГ: открытое овальное окно. Консультация офтальмолога: ангиопатия. Консультация невролога: гипоксически-ишемическое поражение головного мозга, судорожный синдром, синдром угнетения. Дисгенезия мозолистого тела, гипоплазия мозжечка. Хромосомное заболевание? Рекомендована консультация генетика. Консультация генетика: врожденная пневмония. Множественные малые аномалии развития. Хромосомная аномалия? Рекомендовано: хромосомный микроматричный анализ, повторная консультация по результатам. Сдан анализ на полное секвенирование экзома в Медико-генетический центр лаборатории молекулярной патологии Геномед г. Москва (срок исполнения – до 90 рабочих дней). В отделении проводилась антибактериальная, инфузионная, посиндромная терапия.
На 27-е сутки жизни ребенок переведен в отделение патологии новорожденных и недоношенных детей с диагнозом: Врожденная пневмония неуточненная, острое течение, тяжелая форма. ВПР: пролапс мембранозной части трахеи, бронхомаляция среднедолевого бронха справа. Гипоксически-ишемическое поражение головного мозга, судорожный синдром, синдром угнетения. Дисгенезия мозолистого тела. Гипоплазия мозжечка. Множественные малые аномалии развития. Хромосомная аномалия? Открытое овальное окно. Ангиопатия сосудов сетчатки. Осложнение: ДН 2–3 степени. Операция: нижняя трахеостомия. Ребенок признан нуждающимся в оказании паллиативной медицинской помощи по группе 4 решением врачебной комиссии.
В возрасте 2 месяцев и 5 дней ребенок переводится для дальнейшего лечения в отделение детской паллиативной помощи с диагнозом: Основной: Органическое поражение ЦНС, псевдобульбарный синдром, спастический тетрапарез, судорожный синдром. Осложнение: ДН 2–3 степени. Сопутствующий: Хромосомное заболевание, синдром Корнелии де Ланге? Дисгенезия мозолистого тела. Гипоплазия мозжечка. Гипоксически-ишемическое поражение ЦНС. Операция: Нижняя трахеостомия. Для дальнейшего лечения переводится на стационарную паллиативную койку в ГДКБ № 17 по зоне ответственности на основании приказа Минздрава Республики Башкортостан от 17.05.2023 № 963-А «О временной маршрутизации детского населения при оказании медицинской помощи по профилю “паллиативная медицинская помощь детскому населению” в медицинских организациях» на 2023–2024 гг.
Ребенок поступает в паллиативное отделение ГДКБ № 17 в возрасте 3 месяцев и 18 дней
Жалобы при поступлении: на нарушение сознания, дыхания, глотания, сосания, вынужденную позу, ограничение произвольных движений, высокий мышечный тонус конечностей, судороги.
Объективно: Т – 37,7 ºC, ЧД – 42 в мин; SpO2 97 %; ЧСС 145 в минуту, АД 85/45 мм рт. ст. Состояние ребенка тяжелое, стабильное. Тяжесть состояния обусловлена неврологическим дефицитом, дыхательной недостаточностью. Кожные покровы бледно-розовые, теплые на ощупь, тургор мягких тканей сохранен. Подкожно-жировая клетчатка развита удовлетворительно, распределена равномерно. Отеков нет.
Заключение по результатам исследования ДНК методом клинического секвенирования (полное секвенирование экзома)
|
Вариант gh38 |
Зиготность |
Ген |
Транскрипт |
кДНК |
АК замена |
Глубина прочтения |
|
Признаки патогенности в комментарии |
||||||
|
Синдром |
||||||
|
1. Варианты, являющиеся наиболее вероятной причиной заболевания |
||||||
|
Релевантных признаков не обнаружено |
||||||
|
2. Варианты, имеющие один или несколько значимых признаков патогенности |
||||||
|
Релевантных вариантов не обнаружено |
||||||
|
3. Варианты с неизвестным клиническим значением. |
||||||
|
chr14:77027129C>G |
Гетерозиготный |
IRF2BPL |
ENST00000238647 |
c.664G>C |
p.Ala222Pro |
29 |
|
Признаки патогенности варианта: Отсутствует в популяционных БД (EXAC, GNOMAD, GENOMED). Другая информация: Несколько компьютерных алгоритмов предсказывают непатогенность |
||||||
|
chr14:77027734G>A |
Гетерозиготный |
IRF2BPL |
ENST00000238647 |
c.59C>T |
p.Pro20Leu |
44 |
|
Признаки патогенности варианта: Отсутствует в популяционных БД (EXAC, GNOMAD, GENOMED). Другая информация: Несколько компьютерных алгоритмов предсказывают непатогенность |
||||||
|
Заболевания, ассоциированные с геном: Neurodevelopmental disorder with regression, abnormal movements, loss of speech, and seizures (618088), AD |
||||||
|
Рекомендуется сопоставление фенотипа пациента с фенотипом заболеваний, ассоциированных с геном, и обследование родителей для установления происхождения варианта (de novo/наследуемый) |
||||||
|
chr20:32433379G>GT |
Гетерозиготный |
ISXL1 |
ENST00000375687 |
c.1182dupT |
p.lle395fs |
170 |
|
Признаки патогенности варианта: Приводит к сдвигу рамки считывания. Отсутствует в популяционных БД (EXAC, GNOMAD, GENOMED) |
||||||
|
Заболевания, ассоциированные с геном: Bohring – Opitz syndrome (605039), AD |
||||||
|
Рекомендуется сопоставление фенотипа пациента с фенотипом заболеваний, ассоциированных с геном, и обследование родителей для установления происхождения варианта (de novo/наследуемый) |
||||||
Множественные стигмы дизэмбриогенеза: тонкие длинные загнутые ресницы, низкая линия роста волос, гипертелоризм, маленький нос, широкая переносица, короткая уздечка языка, готическое небо, форма подбородка – микрогнатия, короткая шея, глаза не закрывает с обеих сторон, гипертрихоз. Наблюдается периодическая гипертермия центрального генеза до фебрильных цифр. Дыхание самостоятельное через трахеостому, проводится постоянная дотация кислорода со скоростью 1 л/мин. При санации через трахеостому отделяемое из трахеобронхиального дерева слизистое, в умеренном количестве. Грудная клетка: цилиндрической формы, симметричная, эластичность удовлетворительная, при пальпации безболезненная. При перкуссии грудной клетки – звук ясный легочный. Аускультативно в легких дыхание жесткое, хрипы проводные, симметричные с обеих сторон. Гемодинамика стабильная. Тахикардия при гипертермии, ритм синусовый правильный, нормосистолия, тоны приглушены, выслушивается систолодиастолический шум с эпицентром во втором межреберье слева от грудины. Пульс на a. radialis – ритмичный, наполнение удовлетворительное. Микроциркуляция – удовлетворительная, время наполнения капилляров на грудине – 3,0 с, время наполнения капилляров на конечностях – 3,0 с. Язык влажный. Кормление по назогастральному зонду, усваивает в полном объеме. Патологического отделяемого из желудка нет. Живот при пальпации мягкий, доступен глубокой пальпации, перистальтика выслушивается. Печень выступает из-под края реберной дуги на 1 см. Селезенка не пальпируется. Стул самостоятельный кашицеобразный, желтого цвета. Мочеиспускание свободное, моча светло-желтая.
Неврологический статус: сознание с апаллическими нарушениями. Большой родничок практически закрыт. Поза вынужденная, с приведением к туловищу и сгибанием рук, разгибанием ног. Тугоподвижность всех крупных суставов. Черепно-мозговые нервы: глазные щели D=S, зрачки сужены, фотореакция средняя. Взор не фиксирует, периодически глазные яблоки плавают, расходящееся косоглазие. Не глотает, псевдобульбарные нарушения. Слабый сосательный автоматизм. Трахеокануляр. Тонус мышц конечностей высокий с дистоническим компонентом, D = S. Сила мышц конечностей 0–1 балл. Сухожильные рефлексы повышены, D = S. Рефлекс Бабинского положительный с обеих сторон. Спонтанная двигательная активность резко ограничена, при манипуляциях защитные автоматизмы. Менингеальных знаков нет. Голову не держит. В отделении ребенку оказывается паллиативная помощь.
В возрасте 4 месяцев получены результаты генетического исследования (таблица).
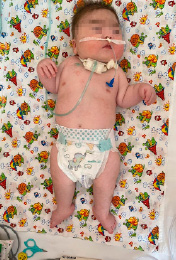
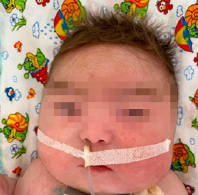
Рис. 1.Характерные клинические проявления у ребенка с СБО (гипертрихоз, гипертелоризм, широкая переносица, микрогнатия)
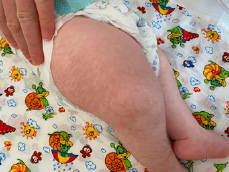
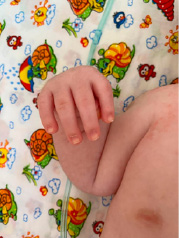
Рис. 2. Характерное положение верхних и нижних конечностей у ребенка с СБО (фиксированное сгибание локтя, двусторонняя контрактура сгибания запястья, контрактура пястно-фалангового сустава большого пальца, гипертония нижних конечностей)
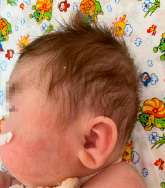
Рис. 3. Характерные клинические проявления у ребенка с СБО (низко посаженные, повернутые кзади уши)
Учитывая характерные клинические проявления: задержка развития нервной системы (очень частые клинические проявления); задержка внутриутробного развития, агенезия мозолистого тела, гипоплазия мозжечка, контрактуры конечностей: фиксированное сгибание локтя, двусторонняя контрактура сгибания запястья, контрактура пястно-фалангового сустава, гипертония нижних конечностей (рис. 2), полные щеки, гипертрихоз (рис. 1), неспособность ходить, рецидивирующие респираторные инфекции, трудности с кормлением, рецидивирующая рвота (частые клинические проявления); гипертелоризм, широкая переносица, микрогнатия, низко посаженные, повернутые кзади уши (рис. 1, 3), судороги (редкие клинические проявления) и результаты исследования ДНК методом клинического секвенирования, выставлен окончательный клинический диагноз: генетическое заболевание, синдром Боринга – Опица.
Заключение
Таким образом, синдром Боринга – Опица представляет собой тяжелое генетическое заболевание. Встречается крайне редко и возникает вследствие генетической мутации de novo. Основными клиническими проявлениями являются множественные врожденные аномалии и пороки развития, характерная форма лица, контрактуры конечностей (поза СБО), гипертрихоз. В представленном клиническом случае подробно описаны клинические проявления и последовательность диагностики у ребенка с синдромом Боринга – Опица. Клинический случай представляет ценность для практических врачей, так как при установке диагноза врач сталкивается с проблемой редкой встречаемости заболевания, выраженной степенью ограничения жизнедеятельности и высоким уровнем младенческой смертности таких пациентов.
Библиографическая ссылка
Санникова А.В., Файзуллина Р.М., Шангареева З.А., Сартания И.Д., Баязитова Г.Р. РЕДКОЕ ГЕНЕТИЧЕСКОЕ ЗАБОЛЕВАНИЕ: СИНДРОМ БОРИНГА – ОПИЦА // Научное обозрение. Медицинские науки. 2025. № 1. С. 22-28;URL: https://science-medicine.ru/ru/article/view?id=1430 (дата обращения: 03.08.2026).
DOI: https://doi.org/10.17513/srms.1430