 science-review.ru
science-review.ru
Заболевания печени, такие как хронический алкогольный гепатит, хронический гепатит вирусной этиологии, цирроз печени, аутоиммунная патология печени, получили широкое распространение среди населения Российской Федерации. Это обусловлено низкой эффективностью современных методов лечения хронических гепатитов, а также малой изученностью молекулярных механизмов этих заболеваний, что затрудняет проведение патогенетической терапии.
Из множества проблем, касающихся иммунной системы печени, мы выделили несколько направлений, наиболее значимых с нашей точки зрения. Одно из направлений поисков заключается в детальном анализе изменений, касающихся иммунного статуса больных хроническими гепатитами. Точные данные о субпопуляции лимфоцитов в печени позволяют направленно удалять наиболее агрессивные субпопуляции, обусловливающие повреждение ткани печени.
I. Классификация хронического гепатита.
Термин «хронический гепатит» известен с 30-х годов 20 века, а его нозологическая самостоятельность определена в 60-е годы. Хронический гепатит – это полиэтиологичное персистирующее повреждение печени, продолжающееся не менее 6 месяцев и сопровождающееся повышением активности трансаминаз и других ферментов, являющихся маркерами воспаления [19,38].
I.1. Классификация, принятая в России.
Классификация хронических гепатитов – постоянно совершенствуется. Международная классификация болезней печени была принята и одобрена Всемирной Организацией Здравоохранения в 1976 году, а затем, с некоторыми добавлениями, включена в Десятый пересмотр Международной классификации болезней (1995 г.) [19, 38]. В основе классификации хронического гепатита положен патоморфологический принцип.
– Аутоиммунный гепатит,
– Хронический гепатит В,
– Хронический гепатит D,
– Хронический гепатит С,
– Хронический вирусный гепатит, не характеризующийся иным образом,
– Хронический неспецифический гепатит,
– Хронический лекарственный гепатит,
– Первичный биллиарный цирроз,
– Первичный склерозирующий холангит,
– Болезнь Вильсона-Коновалова,
– Болезнь печени, вызванная недостаточностью альфа-1-антитрипсина.
В классификацию вносят оценку активности процесса (минимальная, слабовыраженная, умеренная и выраженная), а также стадии заболевания по выраженности фиброза (I – слабовыраженный фиброз, II – умеренный фиброз, III – тяжелый фиброз). В нашей стране по степени активности хронический гепатит подразделяют на персистирующий, активный и лобулярный.
Хронический персистирующий гепатит характеризуется расширением портальных трактов и портальной воспалительной клеточной инфильтрацией. Дольковая архитектура сохранена, фиброз отсутствует или слабо выражен. При хроническом активном гепатите воспалительный инфильтрат распространяется далее на паренхиму печени с нарушением целостности пограничной пластинки, имеются фациальные некрозы, внутридольковые септы, архитектура долек нарушена, но при этом отсутствует узловая регенерация паренхимы [19, 38].
I.1.2. Зарубежные классификации.
Согласно морфологическим изменениям, болезни печени классифицируются как паренхиматозные, гепатобиллиарные и сосудистые [2].
К паренхиматозным поражениям относятся гепатиты, циррозы, инфильтративные поражения печени, объемные образования и функциональные нарушения, сопровождающиеся желтухой. Цирроз печени рассматривается как отдельное заболевание, относящееся к хроническим дегенеративным повреждениям печени, при котором циркуляция крови в печени может быть существенно нарушена, что может привести к портальной гипертензии вследствие обратного тока крови. Гепатиты делятся на вирусные, лекарственные, токсические, ишемические, которые по течению могут быть острые, подострые и хронические. По этиологическому признаку хронические гепатиты рассматриваются как вирусные и невирусные (табл. 1).
Таблица 1
Классификация хронических гепатитов по этиологическому признаку
|
Хронические вирусные гепатиты |
Хронические невирусные гепатиты |
|
Гепатит B Гепатит C Гепатит D (гепатиты А и Е не классифицируются как хронические) |
Лекарственные Аутоиммунные Болезнь Вильсона Недостаточность альфа-1-антитрипсина Алкогольные |
В табл. 2 дается сравнительная характеристика предыдущей и используемой в настоящее время классификаций хронического гепатита.
Таблица 2
Гистологическая классификация хронического гепатита по степени активности процесса
|
Предыдущая классификация хронических гепатитов |
Современная классификация хронических гепатитов |
|
Хронический персистирующий гепатит (гепатит, продолжающийся более 6-ти месяцев, в настоящее время не вызывающий активного повреждения ткани печени, но способный перейти в стадию активного гепатита) |
Хронический гепатит без перипортальных некрозов (поверхностный гепатит) |
|
Хронический активный гепатит (гепатит, продолжающийся более 6-ти месяцев и вызывающий активное повреждение ткани печени) |
Хронический гепатит с перипортальными некрозами (с циррозом или без цирроза) |
|
Хронический лобулярный гепатит |
Хронический гепатит без перипортальных некрозов (умеренный гепатит) |
В настоящее время за рубежом для оценки степени развития фиброза используют классификацию Метавира [22]. F0 – норма, F1 – фиброз, ограниченный портальной зоной, F2 – небольшое количество перипортальных или порто-портальных фиброзных септ с неизмененной архитектурой, F3 – фиброзные септы, мостовидные фиброзы, изменение архитектуры без заметного цирроза, F4 – вероятный или выявленный цирроз. Фиброз может сопровождать любые хронические заболевания печени, при которых имеются гепатоцеллюлярные изменения и воспаление.
I.1.3. Гистологическая характеристика нормальной печени.
Микроархитектоника печени не является гомогенной, она характеризуется наличием вариабельной композиции гексагональных и пентагональных лобулярных единиц, содержащих традиционные портальные триады. Ацинусы печени включены в лобулярные структуры как более мелкие триангулярные физиологические единицы. Вариабельность архитектоники печени более выражена на периферии органа.
При чрезкожной биопсии нормальной печени обычно получают от 2 до 13 фрагментов ткани общей длиной 1,8 +/- 0,8 см и площадью 3,0 – 37,0 см2, которые содержат около от 1 до 18 портальных трактов. Портальные тракты формируют углы печеночной дольки, которая в норме не содержит соединительной ткани и не выделяется отчетливо при гистологическом исследовании. Портальные тракты представляют собой компактные образования в паренхиме печени, окруженные соединительной тканью и включающие в себя, как минимум, две из трех структур, а именно, портальную вену, печеночную артерию и желчный проток. Портальный тракт, включающий все три структуры, называется портальной триадой, а включающий только две структуры – портальной диадой.
В нормальной печени при чрезкожной биопсии около трети всех портальных трактов не содержат портальных вен, 7 % портальных трактов не включают в себя желчный проток, а 9 % портальных трактов не включают печеночной артерии. Портальные тракты, содержащие две печеночные артерии или два желчных протока также являются вариантом нормы, но их число не должны превышать 0,8 +/- 0,5 на 1 мм2 биоптата [12, 28].
Портальная триада, чаще всего обнаруживаемая на периферии печени, содержит один желчный проток, три печеночные артерии, одну портальную вену и один канал Геринга. То есть, в норме портальные тракты представляют собой вариабельные структуры, в которых портальная вена сопровождается печеночной артерией и желчными протоками, но с частой девиацией групповой комбинации этих структур. Портальные диады с двумя структурными компонентами встречаются также часто, как триады. Портальные диады обнаруживаются в любой части печени. Терминальные ветви системы портальных трактов могут быть в виде моно-структур. При патологии печени увеличивается число портальных трактов без желчных протоков, тогда как в нормальной ткани печени это число не должно превышать 7 %.
Минимальный наружный диаметр внутридолькового желчного протока составляет около 13 мм и примерно равен диаметру печеночной артерии. Диаметр портальной вены почти в три раза больше и равен примерно 35 мм. Желчный проток диаметром менее 15 – 20 мкм называют дуктулис, а при еще меньшем диаметре – холангиолой. Диаметр внутридолькового желчного протока колеблется от 20 до 100 мкм, а диаметр септального желчного протока превышает 100 мкм. Внутридольковый желчный проток обычно идет параллельно печеночной артерии. Численное равенство печеночных артерий и желчных протоков является показателем сохранности желчевыводящей системы печени. Уменьшение диаметра желчных протоков по сравнению с диаметром печеночных артерий говорит о наличии деструктивных процессов в желчных протоках, а расширение их – о гиперплазии.
I.1.4. Индекс гистологической активности при хроническом гепатите.
Биопсия печени и последующее гистологическое исследование является необходимым методом диагностики хронического гепатита, который позволяет установить степень активности процесса, тяжесть поражения печени, стадию заболевания.
Гистологическими критериями хронического гепатита являются сочетание воспалительно-клеточной инфильтрации, различных форм гепатоцеллюлярной дегенерации и некроза. При гепатитах токсического и аутоиммунного генеза инфильтрат состоит, главным образом, из лимфоцитов, плазматических клеток и макрофагов.
При хронических заболеваниях печени наиболее выраженные некротические изменения наблюдаются в портальных и перипортальных зонах. Фиброзная ткань формируется внутри и вокруг портальных трактов, обычно сочетаясь с перипортальными некрозами и воспалением. Развивающийся вокруг гепатоцитов фиброз приводит к появлению так называемых гепатоцитарных розеток.
Гепатоцеллюлярный некроз может быть фокальным и включать отдельные клетки или небольшие группы клеток. Сливной некроз наблюдается на месте гибели значительного числа рядом лежащих гепатоцитов. При этом обнаруживают элементы соединительной ткани и клеточного распада, нейтрофилы и макрофаги. Мостовидный некроз возникает при попадании в сливной некроз сосудистых структур. Этот термин обычно обозначает некроз, соединяющий портальные тракты с концевыми разветвлениями вен.
Развитие ступенчатых некрозов свидетельствует о гибели гепатоцитов в пространстве между паренхимой и воспаленной соединительной тканью портального тракта или фиброзной септы. Распространенные ступенчатые некрозы могут приводить к формированию порто-портальных септ.
Для определения степени активности процесса используют индекс гистологический активности по Кноделлю, который исчисляется в баллах.
Индекс гистологической активности по Knodell:
I. Перипортальные +/- мостовидные некрозы (баллы):
– А. Отсутствуют (0),
– В. Легкий ступенчатый некроз (1),
– С. Умеренный ступенчатый некроз (включающий менее 50 % окружности большинства портальных трактов) (3),
– D. Выраженный ступенчатый некроз, (включающий более 50 % окружности большинства портальных трактов) (4),
– Е. Умеренный ступенчатый некроз, сопровождающийся мостовидными некрозами (5),
– F. Выраженный ступенчатый некроз, сопровождающийся мостовидными некрозами (6),
– G. Мультилобулярные некрозы (10).
II. Междольковая дегенерация и фокальные некрозы:
– А. Отсутствуют (0),
– В. Легкие (присутствие ацидофильных телец, баллонирующей дегенерации и/или рассеянных очагов гепатоцеллюлярного некроза в менее чем 1/3 долек или узлов) (1),
– С. Умеренные (вовлечены от 1/3 до 2/3 долек или узлов) (3),
– D. Выраженные (вовлечены более 2/3 долек или узлов) (4).
III. Портальное воспаление:
– А. Отсутствует (0),
– В. Легкое вкрапление воспалительных клеток в менее чем 1/3 портальных трактов (1),
– С. Умеренное (повышенное число воспалительных клеток в 1/3 – 2/3 портальных трактов) (3),
– D. Выраженное (плотные скопления воспалительных клеток в более чем 2/3 портальных трактов) (4).
IV. Фиброз:
– А. Отсутствует (0),
– В. Портальные фиброзы (1),
– С. Мостовидные фиброзы (порто-портальные или порто-септальные) (3),
– D. Цирроз (4).
Степень активности определяется наличием перипортального некроза с мостовидными некрозами или без них (диапазон цифровой оценки от 0 до 10). Далее оценивается интралобулярная дегенерация и наличие фокального некроза (числовая оценка от 0 до 4), а также портальное воспаление (0 – 4). Стадия процесса характеризуется выраженностью фиброза (числовая оценка от 0 до 4) [9, 20].
Гистологический индекс активности 1-3 соответствует хроническому гепатиту с минимальной активностью, что характерно для неспецифического реактивного гепатита. Гистологический индекс активности 4-8 соответствует слабовыраженному хроническому гепатиту, характерному для слабовыраженного алкогольного гепатита. Гистологический индекс активности 9-12 соответствует умеренному хроническому гепатиту, характерному для умеренного алкогольного гепатита. Гистологический индекс активности 13-18 соответствует тяжелому хроническому гепатиту, который соответствует тяжелому хроническому алкогольному гепатиту с мостовидными некрозами [19, 30, 38].
В настоящее время чаще применяют модифицированную гистологическую классификацию, учитывающую изменение архитектоники печени, наличие фиброза и цирроза [Ishak et al., 1995; Knodell et al., 1981] (табл. 3).
Таблица 3
Модифицированный индекс гистологической активности с учетом изменения архитектоники печени, наличие фиброза и цирроза
|
Гистологические данные |
Баллы |
|
Отсутствие фиброза |
0 |
|
Распространение фиброза на несколько портальных областей, без (или при наличии) коротких фиброзных септ |
1 |
|
Распространение фиброза на большинство портальных областей, без (или при наличии) коротких фиброзных септ |
2 |
|
Распространение фиброза на большинство портальных областей при наличии порто-портальных мостовидных некрозов |
3 |
|
Распространение фиброза на портальные области при наличии выраженных порто-портальных (Р-Р) и порто-центральных (Р-С) мостовидных некрозов |
4 |
|
Выраженных мостовидные некрозы (порто-портальные и порто-центральные) с одиночными узлами (неполный цирроз) |
5 |
|
Цирроз |
6 |
В развернутом виде модифицированный индекс гистологической активности включает также характеристику некротических изменений в печени, наличие апоптоза, локального воспаления и воспаления в портальных трактах (табл. 4)
Таблица 4
Модифицированный индекс гистологической активности с учетом некровоспалительных изменений в печени
|
Ступенчатые некрозы |
баллы |
Сливные некрозы |
баллы |
Апоптоз и фокальное воспаление |
баллы |
Портальное воспаление |
баллы |
|
Нет |
0 |
нет |
0 |
Нет |
0 |
нет |
0 |
|
Умеренный фокальный (затрагивает некоторые портальные области) |
1 |
Фокальные сливные некрозы |
1 |
Не более одного очага на 10 полей зрения |
1 |
Умеренное, в некоторых или всех портальных областях |
1 |
|
Умеренный/выраженный фокальный (затрагивает большинство портальных областей) |
2 |
Полоса из трех некрозов в некоторых областях |
2 |
От двух до 4-х очагов на 10 полей зрения |
2 |
Выраженное, в некоторых или во всех портальных областях |
2 |
|
Выраженный (распространяющийся не более, чем на 50 % портальных трактов или септ) |
3 |
Полоса из трех некрозов в большинстве областей |
3 |
От 5 до 10 очагов на 10 полей зрения |
3 |
Выраженное или значительное, во всех портальных областях |
3 |
|
Тяжелый (распространяющийся на 50 % и более портальных трактов или септ) |
4 |
Полоса из трех некрозов плюс редкие порто-септальные мостовидные некрозы |
4 |
Более 10 очагов на 10 полей зрения |
4 |
Значительное, во всех пор-тальных облас-тях |
4 |
|
Полоса из трех некрозов плюс множественные порто-септальные мостовидные некрозы |
5 |
||||||
|
Панацинарные или мультиацинарные некрозы |
6 |
||||||
Таблица 5
Гистологическая диагностика умеренного хронического гепатита и разрешающегося острого гепатита
|
Гистологические данные |
Умеренный хронический гепатит |
Разрешающийся острый гепатит |
|
Портальное воспаление |
+++ |
+ |
|
Лобулярное воспаление |
+ |
+ |
|
Лобулярная гибель гепатоцитов |
++ |
+ |
|
Перипортальная гибель гепатоцитов |
+/- |
+/- |
|
Окраска по Шиффу (+), Диастаза (+) и наличие клеток Купфера |
+/- |
+++ |
|
Железо-положительные клетки Купфера |
+/- |
++ |
|
Персистирующие центролобулярные некрозы |
- |
++ |
Несмотря на модификации первоначального индекса Кноделля, всё же остаются неучтенными некоторые изменения паренхимы печени, в том числе диффузные синусоидальные воспалительные инфильтраты. Модифицированный индекс также не учитывает наличие воспаления и/или повреждения желчных протоков, формирование лимфоидных фолликулов, уровень жировой дистрофии печени, гепатоцеллюлярную дисплазию, аденоматозную гиперплазию, наличие внутриклеточных включений.
Морфологическая характеристика биоптата печени позволяет дифференцировать умеренный хронический гепатит и разрешающийся острый гепатит (табл. 5).
Неспецифический реактивный гепатит.
Термин «неспецифический гепатит» был предложен в 50-х годах 20-го века исследователями F. Schaffner & H. Popper, которые описали воспалительные изменения в печени при заболеваниях желудочно-кишечного тракта.
Неспецифический реактивный гепатит – это вторичный гепатит, вызванный рядом эндогенных и экзогенных факторов, отражающий реакцию печени на какое-либо соматическое заболевание. Он характеризуется умеренно выраженными морфологическими изменениями печени, умеренно выраженными клиническими и лабораторными показателями и доброкачественным течением. Неспецифический реактивный гепатит может выявляться как начальная клинико-морфологическая стадия поражения печени под действием экзогенных факторов, в том числе наркотиков, лекарств и токсинов.
Патогенез заболевания связан с нарушением детоксикационной функции печени по отношению в антигенам и токсинам, поступающим в печень через воротную вену или печеночную артерию
По данным Логинова и Аруина, морфологическая картина печени при неспецифичес-ком реактивном гепатите носит очаговый характер. Наряду с неповрежденными портальными трактами, в некоторых мелких и средних портальных трактах обнаружены анизоцитоз и набухание гепатоцитов, мелкие некрозы паренхимы, окруженные скоплениями макрофагов и лимфоцитов, имеется пролиферация звездчатых ретикулоэндотелиоцитов. Портальные тракты умеренно расширены и инфильтрированы лимфоцитами, гистиоцитами и полимофно-ядерными лейкоцитами, единичными макрофагами, плазматическими клетками и эозинофилами. Пограничная пластинка не затронута. Инфильтрат может распространяться между неповрежденными гепатоцитами в перипортальные участки печеночной дольки. В различных отделах печеночных долек встречаются мелкие очаги некрозов, окруженные инфильтратом из макрофагов, лимфоцитов и нейтрофилов.
В зависимости от локализации воспалительных изменений в печени, выделяют лобулярный и портальный варианты, а по степени активности – персистирующий и активный.
Лобулярный неспецифический гепатит характеризуется очаговыми и сливными некрозами паренхимы, которые расположены вокруг центральной вены. В участках некроза аргирофильная строма разрушена, видны скопления макрофагов, лимфоцитов и нейтрофилов. Портальные тракты отечны, отмечается очаговый или диффузный склероз.
Портальный неспецифический гепатит характеризуется преимущественно отеком и расширением портальных трактов, гистиоцитарной инфильтрацией и некоторым количеством лимфоцитов. Гепатоциты в состоянии белковой и жировой дистрофии, отмечаются некрозы отдельных гепатоцитов.
На ранних стадиях фиброза процесс распространяется только на портальные области. На 2-й стадии процесс распространяется на перипортальную область. На 3-й стадии процесс затрагивает центральную область, формируются мостовидные фиброзы в сторону портальной или центральной области. Вторая и третья стадии являются промежуточными, тогда как 4-я стадия – это цирроз, при котором фиброзы чередуются с элементами регенерации, что обусловливает изменение архитектоники печени.
Стадии воспаления подразделяются на минимальную, среднюю и выраженную. Интерфациальный гепатит характеризуется наличием некрозов и воспалением вокруг портальной области, далее процесс может распространяться на гепатоциты и паренхиму, и далее на портальные тракты [Crawford et al., 1998].
Хронический активный гепатит определяется как длительно текущий прогрессирующий воспалительный процесс, характеризующийся наличием ступенчатых некрозов, лимфоидно-клеточной инфильтрацией портальных трактов, проникающей за пределы пограничной пластинки, образованием коллагеновых волокон и различными видами регенерации гепатоцитов (Логинов и Аруин, 1985).
При холестатическом гепатите в биопсийном материале обнаружена выраженная инфильтрация лимфоцитами, эозинофилами и нейтрофилами, а также пролиферация периферических желчных протоков и следы дегенерации гепатоцитов. Эозинофильная реакция указывает на лекарственный генез заболевания.
Хронический алкогольный гепатит.
Хронический алкогольный гепатит представляет большую проблему для нашей страны. В зависимости от клинической картины и длительности течения заболевания, хронический алкогольный гепатит подразделяется на стадии воспаления, стеатоза и цирроза. Для алкогольного гепатита характерно довольно раннее нарушение структуры долек и переход заболевания в стадию цирроза.
Характерными морфологическими отличиями алкогольного цирроза печени и первичного биллиарного цирроза является преимущественное возникновение фиброза вокруг центральных вен, что завершается формированием мелких узелков. Тогда как при первичном биллиарном циррозе первоначально происходит деструкция желчных протоков за счет гибели эпителиальных клеток, что сопровождается фиброзированием ткани портальных трактов [Подымова, 1998].
При хроническом алкогольном гепатите в стеллатных клетках печени повышается число капель жира, гипертрофируется эндоплазматический ретикулум, клетки начинают синтезировать цитокины, способствующие фиброгенезу. В результате в пространстве Диссе накапливаются волокна коллагена, и нарушается циркуляция между клетками [Han et al., 2001].
В работе Смирнова (2002 г.) показано, что при хроническом алкогольном гепатите в активной стадии нарастает число лимфоидных фолликулов, в центре которых отмечаются скопления макрофагов, иногда содержащих гемосидерин. Однако портальный гемосидероз (то есть наличие гранул гемосидерина в макрофагах, инфильтрирующих портальные тракты) характерен для лекарственного гепатита, что указывает на общность клинико-морфологических проявлений алкогольного и лекарственного гепатита.
В патогенезе ХАГ имеет значение особенности ответа иммунной системы организма на алкоголь. Клетки Купфера продуцируют цитокины, в частности, TNF-a, который вызывает некроз или апоптоз гепатоцитов и непаренхимальных клеток печени. Сигналы гибели клеток проводятся через рецепторы TNF-R1, -R2, CD95 и другие рецепторы апоптоза на мембранах клеток печени.
Жировой гепатоз.
Жировая инфильтрация, жировая дистрофия, стеатоз печени или жировой гепатоз – это синонимы одного заболевания, имеющее характерные клинические признаки, и морфологически характеризующееся отложением нейтральных липидов в гепатоцитах и во внеклеточном пространстве. Если жировая дистрофия сочетается с гепатитом, она выходит на второй план.
Жировая дистрофия печени может иметь различную этиологию, включая ожирение, голодание, эндокринные заболевания, состояние после оперативного вмешательства на желудочно-кишечном тракте, вирусные инфекции, токсические поражения печени. аллергические заболевания, патологию беременности, а также являться стадией развития хронического алкогольного гепатита. Токсические вещества, обусловливающие начало развития жировой дистрофии печени, могут также привести к развитию некротических и воспалительных изменений гепатоцитов. При высокой активности процесса повышена степень фиброзирования ткани печени и переход заболевания в стадию некроза.
Морфологическим критерием жировой дистрофии печени является превышение содержания триглицеридов свыше 10 % от сухой массы. Если более половины гепатоцитов содержат капли нейтрального жира, которые превосходят по величине клеточное ядро, то содержание жира в печени превышает 25 % (Подымова, 1998). Однако классический подход к определению жировой дистрофии подразумевает, что более 50 % гепатоцитов имеют жировые вакуоли [Логинов и Аруин, 1985].
При неалкогольном стеатозе стеллатные клетки печени активируются, в них в 2 раза повышается экспрессия альфа актина. Максимальное скопление активных стеллатных клеток наблюдается вокруг венул, менее – в промежуточной зоне и реже – в портальной зоне. Показана корреляция между активностью стеллатных клеток и портальным и лобулярным воспалением [Cortez-Pinto et al., 2001].
Неалкогольное жировое поражение печени у больных алиментарной формой ожирения может закончиться развитием выраженного фиброза. При ожирении риск развития фиброза связывают в повышением активности одновременно двух факторов – ангиотензина II и трансформирующего ростового фактора-b1 (TGF-b1) [Dixon et al., 2003].
Жировая дистрофия печени обусловливает повышение перекисного окисления липидов в гепатоцитах и лимфоцитах печени. Выделяемые при этом радикалы активируют стеллатные клетки печени, которые, как и клетки Купфера, начинают продуцировать фиброгенные цитокины, в том числе трансформирующий ростовый фактор TGF-b (Transforming Growth Factor). Чем выше степень активности процесса, тем выше степень фиброзирования ткани печени [Shimizu, 2001].
Морфологические изменения в печени при жировой дистрофии характеризуются появлением жировых вакуолей в цитоплазме гепатоцитов. По мере увеличения размера вакуолей они сливаются в одну крупную жировую вакуоль, которая вызывает перемещение ядра на периферию клетки. Разрывы и слияние нескольких клеток приводят к образованию жировых кист. Разрыв жировых кист приводит к формированию липогранулем, которые по составу клеток могут быть макрофагальными или эпителиоидно-клеточными.
В зависимости от размера жировых включений различают мелкокапельную, среднекапельную и крупнокапельную жировую дистрофию, при которых размер капель составляет, соответственно, 2, 11 и 21 мкм [Buntrock et al., 1977]. Крупнокапельная жировая дистрофия обусловливает увеличение размеров гепатоцитов и, соответственно, гепатомегалию. Клиническими симптомами является обратимая портальная гипертензия и гипербилирубинемия.
При жировой дистрофии обнаруживают портальный и центролобулярный фиброз. Портальный фиброз характерен для сопутствующего алкогольного панкреатита, центролобулярный фиброз обнаруживается у всех больных алкогольным гепатитом. Характерным признаком прогрессирования фиброза считается наличие коллагеновых волокон вокруг гепатоцитов.
Отсутствие фиброзных изменений при наличии мелко- и среднекапельной дистрофии печени могут быть обратимы при условии прекращения употребления алкоголя. При крупнокапельной жировой дистрофии с формированием жировых кист и наличием фиброза не происходит регенерация ткани печени [Логинов и Аруин, 1985].
Цирроз печени.
Хронические гепатиты могут иметь различную этиологию и механизмы повреждения ткани печени, однако, конечный результат сводится к прогрессирующему фиброзу и циррозу.
Патогномоничной морфологической картиной цирроза печени является диффузная гиперплазии ткани печени, выражающаяся в развитии паренхиматозных узелков, окруженных фиброзными септами, соединяющими портальный канал и с центральным, и сопровождаемая перестройкой сосудистой архитектоники. Механизмом, обусловливающим развитие паренхиматозных узлов, является активация роста гепатоцитов после некротической гибели паренхиматозных клеток, разделение долек на части фиброзными тяжами, изменение структуры долек вследствие перестройки сосудистого русла. Одним из молекулярных механизмов развития цирроза печени является нарушение активации и дифференцировки стволовых клеток печени, что будет обсуждаться далее.
Цирроз характеризуется потерей функциональной массы печени и функциональной недостаточностью, нарастающей по мере прогрессирования заболевания. Ранние стадии заболевания дают возможность печени компенсировать потерю клеточной массы. Затем наступает период декомпенсации, сопровождающийся снижением числа клеток на единицу объема печени, что заканчивается гибелью организма. Этот период может продолжаться от 1-2-х лет до 20-30-ти лет [Corbin et al., 2003].
Данные биопсии печени отвечают на вопрос о её функциональном состоянии и должны быть дополнены данными биохимии крови и инструментальными методами исследования, включая уровень циркуляции крови в печени, скорость клиренса, метаболизм ксенобиотиков.
Резидуальные гепатоциты должны вырабатывать больше энергии для поддержания активности органа и повышенной регенерации клеток. Нарушенная биоэнергетика клеток приводит к повышенному образованию капилляров в синусоидах печени, что сопряжено с прогрессированием цирроза. Если в норме свободное перисинусоидальное пространство позволяет обмениваться субстратами между гепатоцитами, то в течение фиброгенеза и развития цирроза образуется барьер из белков ЕСМ в перисинусоидальном пространстве, что ограничивает доступ кислорода и нутриентов. Типичные мостовидные фиброзные септы дезорганизуют архитектуру паренхимы. Множественные холлангиофибромы приводят к потере АТФ. Интенсивная пролиферация желчных протоков в перипортальной области обусловливает наиболее выраженное падение АТФ [Corbin et al., 2003].
В клинической практике для снижения степени фиброзирования ткани печени используют декорин, растворимые рецепторы трансформирующего ростового фактора и генную терапию для ингибирования фиброгенного потенциала TGF-b. Одним из естественных ингибиторов TGF-b является ростовый фактор гепатоцитов HGF (Hepatocyte Growth Factor), который активируется при генерации гепатоцитов и резко ослабляет фиброгенез. Токоферол-альфа, ретинол пальмитат и флавоноид силубинин эффективны против окислительного стресса в гепатоцитах и стеллатных клетках печени. Фливоноиды байкалин и байкалеин, сходные по строению с силубинином, снижают фиброгенез в печени [Shimizu, 2001].
I.2. Механизмы апоптоза и некроза и их особенности в клетках печени.
Впервые понятие «апоптоз» было дано в 1972 году учеными Керром, Вилли и Кюри (Kerr, Wyllie, Currie), которые дали четкое определение апоптозу и провели грань между двумя основными типами гибели клеток – апоптозом и некрозом. Если некроз является результатом метаболического коллапса клетки, резкой потери энергии и невозможности дальнейшего поддержание гомеостаза, то апоптоз представляет собой активный процесс, требующий затраты энергии, носящий системный характер, планируемый и контролируемый в соответствии с общим планом развития организма.
Нарушения в реализации программы апоптоза характерны для большинства хронических заболеваний печени, при которых гибель гепатицитов носит массовый характер и приводит к прогрессирующей потере функциональной активности печени.
С точки зрения биохимических процессов в клетке можно выделить несколько типов апоптоза, отличающихся наличием или отсутствием активности определенных классов ферментов, а также сигнальными путями рецепторов апоптоза [12]. Апоптоз условно можно разделить на активационно-индуцированный, глюкокортикоид-индуцированный, Fas-зависимый, TNF- индуцированный, bcl/bcx-зависимый, гранзим- и перфорин-зависимый, каспазо-зависимый и т. д.[26, 38].
Для исследования хронического гепатита наиболее актуальной проблемой является апоптоз, обусловленный потерей мембранного потенциала митохондрий дельта пси мю, а также зависимый от образования реактивных метаболитов кислорода.
Апоптоз является активным генно-регулируемым процессом, морфологически характеризуемым конденсацией хроматина, реорганизацией цитоскелета, потерей контакта с экстраклеточным матриксом, фрагментацией ДНК и цитоплазмы, фагоцитозом изолированных клеточных компартментов без контакта их содержимого с внешней средой, что отличает апоптоз от некроза или других типов распада клеток [18, 41].
Образованные в результате фрагментации цитоплазмы так называемые апоптозные тельца фагоцитируются макрофагами. В тимусе F4/80+ макрофаги имеют малый диаметр и располагаются в корковой зоне рядом с тимоцитами, выходящими в апоптоз, так как обладают высокой скоростью поглощения апоптозных телец [5, 34]. Распознавание и выведение нефункциональных или аутореактивных лимфоцитов также осуществляется макрофагами [4, 29].
Нарушение активации факторов транскрипции, снижение тирозинкиназной активности, ослабление контакта с экстраклеточным матриксом [20, 31] так или иначе влияют на процессы созревания и дифференцировки лимфоцитов, что отражается на количестве клеток, несущих CD3, CD4, CD8, CD25 и другие антигены, а также приводит к изменению плотности и аффинности рецепторов.
Fas-зависимый апоптоз в лимфоцитах является регуляторным механизмом, препятствующим накоплению эффекторных клеток и нерегулируемой продукции цитокинов. Удаление эффекторных CD4+- и CD8+-лимфоцитов после реализации иммунного ответа осуществляется Fas-зависимым путем, который инициируется при связывании лигандом рецептора CD95/Fas/APO-1, реализуется через Ras-сигнальный путь, в котором одной из обязательных ступеней является транзиторный синтез активных форм кислорода [27, 35].
В печеночных Т-лимфоцитах Fas-зависимый апоптоз является одним из двух основных механизмов эффекторной функции цитотоксических лимфоцитов при киллинге клеток-мишеней [8, 27]. Повышение экспрессии CD95 /Fas/APO-1 рецептора на мембране нестимулированного CD4+ лимфоцита повышает вероятность его выхода в апоптоз [19, 26]. По данным [16, 39], апоптоз Т лимфоцитов наблюдается, главным образом, в синусоидах, крайне редко – в портальных трактах, где не превышает 1,1 % от всех присутствующих в них Т лимфоцитов.
В клетках печени сигнальные пути гамма-интерферона и интерлейкинов (IL-6/STAT3 и IL-4/STAT6) взаимодействуют друг с другом. STAT3 ингибирует эффекты гамма-интерферона и повышает экспрессию анти-апоптозного белка Bcl-2 [23, 34]. На биопсийном материале больных хроническим гепатитом показана экспрессия белка Bcl-2 в портальных и интралобулярных лимфоцитах и холлангиоцитах желчных протоков, где этот белок регулирует рост эпителиальных клеток [19, 32]. Сигнальный путь IL-4/STAT6 играет ведущую роль при экспериментальных гепатитах, вызванных митогенными факторами и в клинике аутоиммунных гепатитов [17, 23].
Сигнальный путь STAT6 также регулирует экспрессию IL-13 и его рецептора Ra2, которые рассматриваются как факторы фиброзирования при хронических гепатитах [15, 24].
Одним из специфических механизмов апоптоза в печени является способность гидрофобных желчных кислот вызывать повреждение клеток печени при холестазе. При высокой концентрации желчные кислоты вызывают некроз, а при низкой – апоптоз. При этом билирубин, как акцептор свободных радикалов, защищает клетки печени от окислительного стресса [3, 40]. Желчные кислоты повышают экспрессию рецептора семейства фактора некроза опухолей, так называемого TRAIL-R2/DR5 (TNF-Related Apoptosis Inducing Ligand), на клетках печени, а также повышают чувствительность гепатоцитов к апоптозу. При холестазе TRAIL и его рецептор TRAIL-R2 прямо индуцируют апоптоз гепатоцитов и непаренхимальных клеток печени [18, 21].
По данным [6, 32] апоптоз основной массы Т-лимфоцитов наблюдается в синусоидах и составляет от 4,6 до 6,2 %. Крайне редко апоптоз Т- лимфоцитов наблюдается в портальных трактах, где не превышает 1 % от всех Т клеток. В нормальном состоянии ткани печени апоптоз лимфоцитов является физиологическим процессом, способствующим удалению аутореактивных клеток или антиген-специфических клеток после реализации иммунного ответа на данный антиген.
В нормальной ткани печени нет (или минимальна) экспрессия гранзима и перфорина – гранулированных сериновых протеаз, являющихся основным оружием цитотоксических лимфоцитов, которые внедряют гранулы протеаз в клетки – мишени и вызывают их гибель. При хроническом гепатите показано увеличение экспрессии гранзима и перфорина параллельно с увеличением воспалительной реакции в паренхиме печени. При этом показано, что положительными по этим протеазам является небольшое число внутрипеченочных лимфоцитов и большинство клеток Купфера. Таким образом, именно клетки Купфера осуществляют гранзим-зависимый и перфорин-зависимый апоптоз гепатоцитов [28, 31].
Показана корреляция между числом клеток, выходящих в апоптоз, и степенью лобулярных некрозов [1, 14, 30]. Следует особо отметить, что на гепатоцитах, восстанавливающихся на месте некрозов, экспрессируется повышенное число Fas/CD95/Apo-1-рецепторов и белка Вах, который является про-апоптозным белком, локализованным на мембране митохондрий. Частота апоптозных клеток коррелировала с экспрессией Вах и степенью инфильтрации воспалительными клетками. То есть, апоптоз играет важную роль в массивной гибели гепатоцитов при хроническом гепатите.
При некоторых заболеваниях печени показано наличие уникального механизма защиты гепатоцитов от апоптоза, когда гепатоциты продуцируют растворимый Fas-антиген (sCD95), который блокирует сигнальные пути Fas-индуцированного апоптоза и, таким образом, защищает печень от потери функциональной массы клеток [7, 38].
В модельной системе на крысах с использованием магнитно-резонансной спектроскопии показано, что уровень внутрипеченочного АТФ прогрессивно падает с развитием цирроза печени [22, 37]. Снижение уровня АТФ в печени коррелирует с потерей массы гепатоцитов. На поздних стадиях цирроза потеря АТФ идет пропорционально потере функциональной массы печени.
Интерфациальные некрозы – это тип гибели гепатоцитов, обусловливающий постепенное исчезновение массы печени и стирание границы между долькой и портальным трактом. Их наличие показано при аутоиммунных гепатитах, стеатозе, вирусных гепатитах, первичном биллиарном циррозе. Пусковым механизмом гибели гепатоцитов является их непосредственный контакт с лимфоцитами, которые взаимодействуют с рецепторами апоптоза на гепатоцитах или выстреливают гранулы сериновых протеаз, что морфологически проявляется типичными признаками апоптоза – сморщиванием цитоплазмы, ядра, фрагментацией клетки и поглощением апоптозных частиц макрофагами [9, 11].
При хроническом гепатите С активация каспаз идет параллельно с развитием воспалительного процесса. Ингибиторы каспаз уменьшают скорость гибели гепатоцитов, но при этом создаются условия для персистирования вируса в ткани печени [6, 29, 36].
I.3. Аутоиммунные повреждения паренхимы печени.
Аутоиммунные реакции характеризуются продукцией антител, реагирующих со структурными компонентами собственных клеток и тканей. При аутоиммунитете образуются эффекторные Т-лимфоциты, распознающие собственные пептиды. Активация Т-лимфоцитов происходит как неспецифически, так и перектрестно-реагирующими антителами. Аутоиммунитет делится на системный и органоспецифический. К аутоиммунным поражениям печени относятся первичный биллиарный цирроз, склерозирующий холангиит, аутоиммунный хронический гепатит.
В печени происходит потеря толерантности к собстенным антигенам, что может начинаться без видимых клинических причин. Однако у таких больных в анамнезе практически всегда была какая-либо вирусная инфекция. Вирусные инфекции, включая вирус гриппа, могут служить пусковым механизмом развития аутоиммунитета. Механизмом инициации аутоиммунных реакций вирусами может быть продукция так называемых антиген-зависимых неспецифических иммуноглобулинов, продукция которых идет параллельно с синтезом антител, но превышает их на порядок [Сидорова, Агаджанян и др.]. У больных первичным циррозом печени повышен уровень иммуноглобулинов всех классов, что указывает на поликлональную активацию лимфоцитов.
В последнее время активно обсуждается вопрос о генетической предрасположенности некоторых лиц к аутоиммунной патологии печени. Например, антимитохондриальные – антитела, которые обнаруживаются у 95 % больных первичным биллиарным циррозом, также находят и у практически здоровых лиц. Диспансерное наблюдение за этими пациентами показало, что у них рано или поздно начинает развиваться поражение печени [7, 32].
Для уточнения механизма патологии следует обращать внимание на такие факторы, как пол пациента, так как первичный биллиарный цирроз чаще развивается у женщин (> 90 % случаев). Необходимо учитывать ассоциацию с другими аутоиммунными заболеваниями, включая ревматоидный артрит, исследовать систему HLDA (Human lymphocyte Differential Antigenes).
ПБЦ характеризуется селективной деструкцией внутрипечёночных протоков и формированием гранулем [13, 39]. В портальном тракте, особенно вокруг межлобулярных желчных протоков, накапливаются CD3+CD57+ NK клетки. Эти клетки аутоагрессивны и регулируют изменение локального иммунитета в перидуктальном микроокружении. Гистологически выявляются деструктивные холлангиты, холестаз, дегенерация гепатоцитов, деструкция малых желчных протоков, пролиферация мелких желчных протоков, вокруг которых наблюдается инфильтрация лимфоцитами, эозинофилами и плазматическими клетками. В портальной зоне формируются гранулемы. Архитектура печени нарушена за счет появления фиброзных септ и регенерирующих модулей [4, 18, 33].
У больных ПБЦ значительно повышены показатели щелочной фосфатазы сыворотки и гамма-глютамилтранспептидазы, повышение которых является диагностическим критерием ПБЦ, подтверждающим холестаз. АЛТ и АСТ повышены, но значительно менее выражено. Обычно повышается общий билирубин, холестерин, ТГ и ЛПНП, чаще в начале процесса и при декомпенсации [1, 12]. IgM повышен в 2-3 раза, IgG и IgA обычно в норме. Повышение IgM отражает митогенную активацию В-лимфоцитов как следствие продукции аутоантител.
Гипергаммаглобулинемия и аутоантитела, которые являются диагностичеким фактором при ПБЦ, обнаруживаются и при верифицированном гепатите С. Классическая триада сопутствующих заболеваний, характерная для ПБЦ, то есть синдром Съёгрена, тиреоидит и целиакия, в равной мере встречаются как осложнения при хроническом гепатите С, а аллель HLDA B35, наиболее часто тестируемая у больных ПБЦ, является плохим прогностическим признаком при хроническом гепатите С и ВИЧ инфекции [17, 24].
Из истории аутоиммунитета: известно, что в 1954 году Медавар (Medawar P.) открыл, что избыток антигена тормозит рост антиген-специфических клонов лимфоцитов. В 1969 году Бернет выдвинул теорию клональной селекции (Burnet F.M. «The clonal selection theory of acquired immunity»), в которой, в частности, говорилось о том, что соматические мутации во взрослом организме приводят к появлению так называемых «запрещенных клонов» лимфоцитов, реагирующих со своим антигеном. В 1971 году Вайгл и Аллисон (11,32) показали, что низкое содержание в крови «своего» антигена может элиминировать Т-лимфоциты, сохраняя при этом предшественники В-лимфоцитов, способных распознавать этот «свой» антиген.
Исторические справки имеют непосредственное отношение к патогенезу хронических поражений печени. Дело в том, что печень в нормальном состоянии призвана быть толерантной к антигенам, особенно пищевого происхождения. При хронических реактивных гепатитах, алкогольных, лекарственных нельзя исключить аутоиммунного компонента в патогенезе, хотя он может и не играть ведущей роли. Эти рассуждения касаются механизма гибели гепатоцитов и потери массы функционально активных клеток печени, что приводит к печеночной недостаточности. Помимо перечисленных нами ранее механизмов апоптоза и некроза, в ряде случаев гибель гепатоцитов может носить аутоиммунный характер, например, при интерфациальном некрозе.
В патогенезе первичного биллиарного цирроза лежат аутоиммунные нарушения. Эта нозология сопровождается выраженными изменениями фенотипа лимфоцитов, изменением экспрессии молекул адгезии и усилением апоптоза [2, 13]. В настоящее время рассматривается ведущая роль в патогенезе ПБЦ антимитохондриальных антител и gp-210 реактивного комплекса, связывающего комплекс ядерных пор [16, 22].
При ПБЦ повышена экспрессия рецептора LFA-1 на CD4+ Т-лимфоцитах, что приводит к преимущественной мобилизации в печень Т-хелперов первого порядка (Th1) [3, 8, 22]. Цитотоксические лимфоциты и Th1 клетки имеют эпитопы сходной аминокислотной последовательности, что способствует распознаванию чужеродного антигена аутоспецифическими Т-лимфоцитами. При этом повышается относительная эффективность презентации антигена Т-лимфоцитам [4, 18, 24].
Аутоиммунная патология часто сопровождается нодулярной регенеративной гиперплазией печени, которая сопровождается портальной гипертензией в отсутствие цирроза печени. Одним из методов лечения является прием урсодезоскихолевой кислоты, которая существенно улучшая функциональное состояние печени [1, 16, 33].
При изучении хронического гепатита, обусловленного аутоиммунной патологией, наиболее актуален вопрос о механизмах активационно-индуцированного апоптоза. Основной задачей этого типа апоптоза является выведение из организма зрелых Т-лимфоцитов, которые были активированы через TCR/CD3 комплекс или ко-рецепторы CD4 или CD8, но в отсутствие специфического антигена не могут стать эффекторами [7, 19]. Активационно-индуцированный апоптоз может быть при распознавании рецептором CD4+ Т-лимфоцита растворимого комплекса, состоящего из антигенного пептида и молекулы MHC II класса, который в норме должен быть представлен на мембране антиген-презентирующей клетки, что сопровождается дополнительными сигналами от других мембранных рецепторов [9, 16].
В настоящее время показано, что аутоимунные заболевания могут быть не только следствием потери центральной толерантности, но и результатом повышения экспрессии генов RAG1 и RAG2. Гены RAG ответственны за активацию рекомбинации генов. Экспрессия генов RAG1 и RAG2 повышена на аутоагрессивных клетках. Рецептор CD40, который широко экспрессирован на клетках иммунной системы, способен регулировать экспрессию этих генов и, таким образом, появление аутоагрессивных клонов [4, 15].
I.4. Механизмы регенерации ткани печени.
I.4.1. Общие представления о стволовых клетках.
Регенерация ткани печени осуществляется за счет активации коммитированных стволовых клеток. Присутствие предшественников печеночных предшественников подтверждено для человека, в частности в тех случаях, когда развиваются гепатоцеллюлярные карциномы и холангиокарциномы на фоне хронических вирусных гепатитов [2, 9, 13].
Необходимо сразу же четко определить термины и понятия. Выделяют несколько типов стволовых клеток (СК) и иерархию этих типов по степени недифференцированности: тотипотентные, плюрипотентные, линейно-рестриктированные СК.
Общим для всех типов СК является отсутствие дифференцировки, наличие характерного, уникального для СК фенотипа, способность к неограниченному самовоспроизводству и образованию более дифференцированных типов клеток.
Клетки эмбрионов до 16-клеточной стадии относятся к тотипотентным СК, или тотально-потентным. Они 1) способным к неограниченной и недифференцированной пролиферации in vitro, 2) могут давать начало всем типам клеток, включая клетки, поддерживающие развитие эмбриона, 3) могут дать начало новому жизнеспособному организму или регенерировать любую его часть. Они имеют нормальный набор хромосом, высокий уровень теломеразной активности (определяющей бессмертие СК), экспрессируют клеточные поверхностные маркеры, характеризующие уникальный фенотип СК. После недифференцированной пролиферации в течение 4 – 5 месяцев, эти клетки поддерживают потенциал к формированию трофобласта и производству всех трех эмбриональных зачатков.
На стадии бластоциста образуется группа клеток, так называемая внутренняя клеточная масса, состоящая из плюрипотентных стволовых клеток. Они способны воспроизводить любые клетки трех основных типов тканей – эндодермы, мезодермы и эктодермы. Однако ни плюрипотентные СК, ни тем более их производные, не способны к образованию целого эмбриона при имплантации, так как не могут сформировать плаценту или другие экстраэмбриональные ткани. Эти клетки положительны по щелочной фосфатазе, антигену CD34, и т.д.
Более специализированные СК, способные давать только одну или несколько типов тканевых клеток (печеночные клетки, мышечные, нервные, клетки крови, и т.д.), т. е. предрасположенные к выполнению определенных функций, относятся к линейно-рестриктированным СК. Организм взрослого человека содержит множество таких СК, отвечающих за воспроизводство утраченных клеток организма. Для них характерна потеря антигена CDw90 и приобретение рецептора CD38+. Одновременно появляются линейно-специфические антигены.
Во взрослом организме эмбриональные СК остаются в гонадах в виде эмбриональных половых клеток – предшественников гамет. Они сохраняют все свойства эмбриональных СК, в том числе теломеразную активность. Эмбриональные СК способны дать 210 типов клеток организма человека. Упрощенная схема современного представления о стволовых клетках приведена на рисунке.
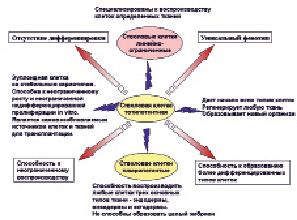
Классификация и функциональная активность различных популяций стволовых клеток
I.4.2. Бессмертие стволовых клеток.
Обычные клетки дают не более 50 поколений, после чего их деление прекращается. Это определяется теломерами – сегментами ДНК, играющими роль в развитии и делении клеток. На теломере расположен участок, на который садится считывающая машина при копировании ДНК при делении клетки. При каждом делении хромосома становится короче на этот сегмент, так как он не копируется. Когда укорочение достигает критической длины, дальнейшее деление клетки становится невозможным.
Опухолевые клетки содержат ген теломеразы, восстанавливающий длину хромосом до естественного уровня. СК сначала превращаются в более коммитированные, линейно-ограниченные СК и далее в специализированные, потерявшие теломеразную активность.
Одним из механизмов ограниченной возможности печени к регенерации при циррозе является укорочение теломеры в гепатоцитах, что ограничивает число клеточных делений. Длина теломеры в гепатоцитах при циррозе достоверно уменьшена по сравнению с нормальной тканью, что не зависит от этиологии заболевания и возраста пациента. При этом длина теломеры нормальна в лимфоцитах и стеллатных клетках печени из зоны фиброза. В подавляющем большинстве случаев укорочение теломеры строго коррелировало с повышением бета-галактозидазной активности, которую рассматривают как маркер старения [6, 12]. Изменение архитектоники печени при хроническом гепатите, переходящем в цирроз, сопровождается укорочением теломер и реактивацией теломеразной активности в клетках регенеративных узлов. Этот процесс является характеристикой, начинающейся гепатокарциномы [14, 23].
I.4.3. Стволовые клетки печени.
В печени найдены компартменты, в которых расположены стволовые клетки. Это особые структуры в желчных протоках. Эти клетки начинают экспрессировать белки MDR1, MRP1, MRP3 при некоторых хронических заболеваниях печени. В нормальной печени экспрессия белков MDR3, BSEP, MRP2, MDR1 ограничена каналикулярной мембраной гепатоцитов и холангиоцитов. Экспрессия MRP3 ограничена эпителиальными клетками желчных протоков и центрилобулярных гепатоцитов. При вирусных гепатитах, как и при ВИЧ-инфекции, значительно возрастает экспрессия MDR1, MRP1, MRP3, особенно на фоне выраженных некротических поражений паренхимы печени. Эти белки играют роль в дифференцировке печеночных предшественников и репарации ткани печени [5, 18, 33].
Бипотентные предшественники, способные к дифференцировке в гепатоциты и биллиарный эпителий, обнаружены у больных хроническим гепатитов С. Для этих так называемых «овальных» клеток характерна экспрессия некоторых маркеров, в том числе лимфотоксина-бета (LT-b) семейства фактора некроза опухолей, p-глютатион-трансферазы, М2-пируват-киназы и т.д. Лимфотоксин-бета повышается у больных хроническим гепатитом С в несколько раз, он экспрессируется на печеночных предшественниках, клетках воспалительного инфильтрата и малых портальных гепатоцитах, и играет роль в регенерации ткани печени [17, 27]. Показана прямая зависимость между числом «овальных» клеток и уровнем повреждения ткани печени при хроническом гепатите С [19, 28]. Повышение уровня пролиферации печеночных предшественников прямо коррелирует с повышением риска развития гепатоцеллюлярной карциномы при циррозе печени [10, 21].
Другими маркерами стволовых клеток печени являются: рецептор фактора стволовых клеток c-kit, кластер дифференцировки CD34, цитокератины СК18, СК19 и т.д. [27, 33]. При хронических гепатитах наблюдается так называемая «типичная пролиферация протоков», обусловленная активацией стволовых клеток. При острых гепатитах пролиферация идет, главным образом, за счет зрелых гепатоцитов, при хронических гепатитах значительный вклад в процессы репарации вносят печеночные предшественники. При некоторых гепатитах, особенно длительно и активно текущих, когда потенциал зрелых гепатоцитов резко падает, регенерация ткани идет только за счет стволовых клеток печени. Они дают начало клеткам протоков и, частично, малым гепатоцито-подобным клеткам [23, 28].
При регенерации печени показано наличие нескольких типов клеток – предшественников на основе проведенного ультраструктурного анализа. Предшественники новых клеток протоков приобретают свойства полярных гепатоцитов в результате многоступенчатого процесса, который включает в себя миграцию новых клеток в паренхиму и распределение их в базальной мембране [12, 32].
Аутоиммунный гепатит 1-го типа сопровождается повышением активности стромальных предшественников и коммитированных стволовых клеток, тогда как при первичном биллиарном циррозе, наоборот, активность предшественников резки снижена [13, 33].
I.4.4. Стволовые клетки как новое направление в терапии хронического гепатита.
Целью трансплантации СК является восстановление функции организма, в котором имеется дефект органа или ткани. Основная цель этого направления – это индукция клеточного деления и пролиферации СК. В настоящее время основными источниками СК для пересадки являются 1) пуповинная кровь и 2) выделение из взрослого организма с помощью флюоресцентно-активационного сортера линейно-рестриктированных стволовых клеток на основании их специфических поверхностных маркеров.
Другим методом получения стволовых клеток является перенос ядер соматических клеток. Считается, что любая соматическая клетка взрослого организма может быть слита с денуклеатированной яйцеклеткой и перепрограммирована на недифференцированный, тоти-, или плюрипотентный статус. Ожидается создание нового метода, включающего в себя как получение примордиальных СК, так и перенос соматических ядер.
Примордиальные клетки являются универсальными донорами, а замена ядер позволит избежать иммунного конфликта. Предполагается, что, как и в случае с трансплантацией органов, можно будет использовать одну донорскую ДНК как группового универсального донора. При хроническом гепатите пересадка стволовых клеток позволит восстановить массу гепатоцитов и функциональную активность печени.
При первичном биллиарном циррозе печени существенно снижена активность коммитированных стволовых клеток в печени и костном мозге, тогда как при аутоиммунном гепатите 1-го типа повышена активность гемопоэтических предшественников, повышено число CD34+, CD34+/CD38+ и CD34+/CD38- клеток [13, 33].
Стволовые клетки печени эндодермального происхождения могут давать начало гепатоцитам и клеткам желчных протоков [35, 41]. По данным (B. Petersen, Science, 1999), костномозговые стволовые клетки способны дифференцироваться в основные клетки печеночной ткани, что указывает на возможную роль костного мозга в поддержании и восстановлении функции печени. При специальных условиях эти клетки дифференцируются в большинство типов функционально-активных клеток печени. Часто после повреждения печень способна генерировать новые гепатоциты, в этом процессе принимают участие стволовые клетки костномозгового происхождения.
Библиографическая ссылка
Нурмагомаев М.С., Магомедова З.С., Каграманова З.С. ЛИТЕРАТУРНЫЙ ОБЗОР: ХРОНИЧЕСКИЕ ГЕПАТИТЫ В КЛИНИКЕ ВНУТРЕННИХ БОЛЕЗНЕЙ // Научное обозрение. Медицинские науки. 2016. № 5. С. 77-91;URL: https://science-medicine.ru/ru/article/view?id=932 (дата обращения: 27.06.2026).